TRANSIT2 Overview
Transit2 is a python-based software system that combines statistical algorithms for analyzing TnSeq data (from sequencing transposon-mutant libraries). It has GUI (graphical-user interface) to make it easy for users to process their data, conduct essentiality analyses, and visualize results. Transit routines can also be run from the command line. It works in Linux, MacOS, and Windows. Transit requires various python packages to be installed, as well as R (a statistical program that Transit calls for some analyses).
Transit2 has two main phases:
a pre-processing phase (called TPP) which analyzes raw sequencing data (fastq files) and extracts transposon insertion counts at genomic coordinates (i.e. TA dinucleotides, for the Himar1 transposon), and
statistical analysis methods (using Transit GUI or command line) - gumbel, HMM, resampling, ANOVA…
The statistical analyses are focused on identifying genes that are essential or conditionally-essential, and quantifying the statistical significance of these. Although Transit is primarily designed for Himar1 transposon libraries, some of the methods can also be applied to other transposons, like Tn5.
NEW in Transit2: The GUI and command-line versions of Transit have been overhauled to now take combined_wig files (and corresponding metadata files), instead of individual .wig files. Thus, after running TPP on all the individual samples, the multiple .wig file are combined together in a single combined_wig file with multiple columns. This makes it much more convenient to manage and analyze large datasets.
Transit2 represents a complete overhaul of the software and GUI interface. The commands and flags have changed, and the python API (internal data structures and functions) have changed substantially. The new GUI is more integrated for ease-of-use, has some new features (like ANOVA and ZINB), and will make it easier to add new features in the future.
Note
For previous users of Transit who prefer backwards compatibility, we will continue to maintain the original version of Transit (Transit1). See the documentation and installation instructions
Typical Workflow
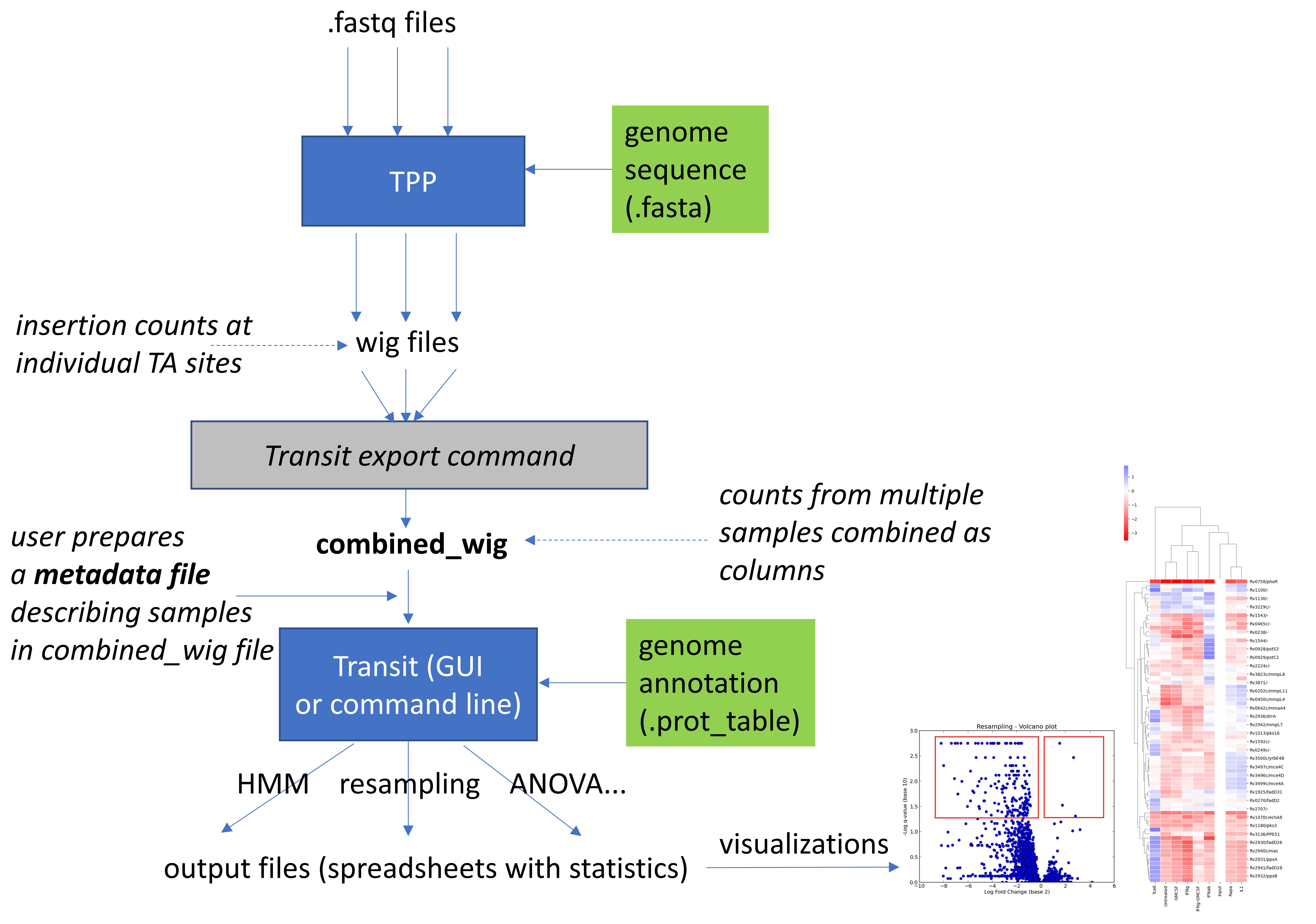
A typical workflow follows 3 phases, illustrated below.
First, the user runs TPP on raw sequence files (.fastq) to produce .wig files with insertion counts at coordinates of TA sites for each sample.
Second, the user runs a command to merge the .wig files into a single combined_wig file, and prepares a corresponding metadata file describing the samples.
Third, the user runs various statistical analysis methods in Transit, using the combined_wig and metadata files as inputs.
Note
- Sequencing Recommendations
Read Length: at least 75 x 75 bp paired-end reads
Sequencing Depth: 5-10 million reads per sample (for M. tuberculosis libraries) to get sufficient dynamic range of counts
Ideally, 3 replicates per condition
Most of the analyses in Transit produce an output file in tab-separated format that can be opened as a spreadsheet. For example, ‘resampling’ generates an output file that lists the data (mean insertion counts in 2 conditions being compared) and statistics (log-fold-change, p-value, adjusted p-value) for each gene in the genome. The user can then view this output file (in the GUI, or open it in a program like Excel) to examine genes predicted to be conditionally essential.
TPP (Transit Pre-Processor)
TPP is a tool for analyzing raw sequencing data (.fastq files) and tabulating insertion counts at TA sites. TPP generates .wig files as output, which simply contain a pair of columns: 1) coordinates of TA sites, and 2) counts of insertions observed at each site.
If Himar1 Tn libraries are prepared according to standard protocols, such as (Long et al. 2015), then the sequencing reads will have a prefix correspond to the transposon terminus, followed by genomic DNA (along with template barcodes in read 2).
TPP removes the prefix and maps the genomic portion of the read into the genome (using BWA, Li and Durbin, 2009). It also reduces counts to unqiue templates at each site, which reduces noise.
For diagnostics, each TPP run also generates a .tnseq_stats output file which contains various metrics, such as the density (percent of TA sites with insertions), total reads, mapped reads, and the NZmean (mean counts at non-zero sites). It also reports statistics on how many reads were rejected because they contained primer, adapter, or vectors sequences, which can be useful for diagnosing problems with the library.
For Tn5 datasets (e.g. generated the MmeI restriction enzyme, van Opijnen et al. (2015)), the differences in running TPP are: 1) there is no prefix, since the transposon terminus does not appear in the reads, and 2) the .wig files contain coordinates and counts for all sites in the genome, since Tn5 is not restricted to insertions at TA sites (unlike Himar1).
Combined_wig and Metadata Files
Once the .wig files are created, they are combined into a combined_wig file. This is a new step in Transit2. Multiple .wig files are combined into a single combined_wig file using the ‘transit export combined_wig’ command on the command line. Users then prepare a metadata file describing each of the samples (as a spreadsheet, e.g. in Excel, which is then saved in tab-separted format). Most commonly, there will be several replicates associated with each condition. The analysis methods in Transit all have been redesigned to take combined_wig and metadata files as inputs, which simplifies things when working with large datasets.
By default, the insertion counts in each dataset (.wig file) are normalized by TTR (Total Trimmed Read-count) when they are combined in a combined_wig file. This facilitates comparing insertion counts at individaul TA sites across samples. If alternative normalization (or none) is desired, this can be specified using a flag in the ‘export combined_wig’ command.
It is often useful to examine at the pattern of insertions in conditionally-essential genes in combined_wig files to confirm what the statistical analyses label as “significant” genes, e.g. to ensure that the result is not biased by outliers (high counts) at individual sites, but rather that apparent differences in counts between conditions are reflected as a consistent trend over multiple TA sites in a gene. This is a recommended practice.
Once a combined_wig file is prepared, it can be used to assess data quality. There are two methods available. First, there is a tnseq_stats command, which will calculate various metrics for each sample, include saturation (density, percent of TA sites with non-zero insertions), mean read count (NZmean), as well as skewness and other statistics of the read-count distribution (for diagnostic purposes). This can be run at the command-line and in the GUI. Also, plots of read-count distributions can be generated for selected samples in the GUI (again, helpful for identifying highly skewed samples). A discussion about skewed samples, the problems they cause, and what to do about them can be found here (Quality Control).
One can also evaluate and compare samples by making scatter plots and pairwise correlation plots, to get a preview of how samples are related to each other.
Genome Annotations (.prot_tables and .gff files)
The annotation of a genome contains information about genes, such as coordinates, strand, locus tag, gene name, and functional description. Transit uses a custom format for annotations called prot_tables, e.g. H37Rv.prot_table. Prot_tables are tab-separated text files containing the information on each gene, such as coordinates, strand, ORF id, and gene name.
In many cases, users might often obtain annotations for their genome in .gff (or .gff3) file format, such as downloaded from NCBI. .gff files contains essentially the same information about genes. However, there is a bit more flexibility in the .gff file format (especially in the tags used in the right-most column), and the information about genes is not always encoded in a uniform way, making it difficult to use arbitrary .gffs for analyses in Transit. Therefore, there is a simple procedure in Transit to convert a .gff file to .prot_table format (see instructions for converting .gff files to .prot_tables). This step only has to be done once, and then the .prot_table can be used for all subsequent analyses in Transit.
GUI
Here is a screenshot of the new GUI in Transit2:

Basic walk-through:
You start by loading 3 input files (in succession): combined_wig, metadata, and annotation. This will populate the upper panel with individual samples, and the middle panel with conditions.
Next, you can evaluate certain samples by selecting them and then choose an Action from the ‘Select Tool’ dropdown box above the samples panel, such as displaying track views, making scatter plots, examining chromosomal bias via LOESS plots, and showing plots of read-count distributions (for quality control).
Next, you can select an analysis method from the menu (under Methods->himar1), such as gumbel, HMM, resampling, ZINB… This will bring up a corresponding parameter panel on the right. You might need to select specific samples or conditions to analyze. You can usually use the defaults for the other parameters.
Then you hit the Run button. You should be able to monitor progress via the Progress Bar.
Status updates and various messages will be displayed in the Message Bar at the bottom of the GUI window. Important: The full list of messages, including error messages will be printed on in the console window from where you started Transit. Check these message if anything goes wrong.
Usually, an analysis method will generate one or more output files. These are typically text files in tab-separated format (which could be opened as spreadsheets in Excel). The file will get populated into the Results Table (bottom pannel).
If you select an output file in the panel, it will provide a dropdown with Actions you can perform, including displaying the file as a table (or figure, if it is an image, such as volcano plots, heatmaps, etc). Some output files have customized Actions, such as making a volcano plot from output of resampling, or making a heatmap from the output file after running ANOVA or ZINB.
One of the most common and important Actions (‘Select Tool’ dropdown under Results Files) is to perform is Pathway Enrichment Analysis on the genes found to be significant by one of the other analyses (e.g. gumbel, hmm, resampling, ANOVA). Most of these output files have a column with adjusted P-values, and significant “hits” are usually defined as genes with Padj<0.05. If you have more than about 20 hits, you can use Pathway Enrichment Analysis to determine whether they share functional similarities. There are several systems of pathways available, including COG categories and GO terms.
Pre-Processing Tasks
When you first start the Transit GUI, you load up your TnSeq dataset (combined_wig file). Before running any statistical analyses, the first thing you will probably want to do is explore the individual samples, their relationships, and data quality. Most of these steps can be performed by clicking on a sample in the samples menu and selecting an action from the drop-down list, or by choosing one of the items from the Pre-Processing menu. These steps can also be done at the command line.
generate a tnseq_stats table (under Pre-Processing menu) summaring key statistics and metrics for each individual sample (including saturation, skewness, etc)
generate a LOESS plot (drop-down list) to see whether the mean read count variest significantly across the genome (chromosomal position bias; M. tuberculosis samples typically do not show a substantial bias)
examine distributions of read-counts and QQ-plots (select ‘Quality Control’ in drop-down list) to check for highly skewed samples
look at a Track View (drop-down list) for one or more samples that shows insertions (vertical bars) at TA sites in a target gene or locus
compute a gene means spreadsheet, with the mean insertion count in each gene in each condition (Pre-Processing->Gene Means) - this generates a helpful spreadsheet from which one can make barcharts showing how the (normalized) insertion counts vary across conditions for genes of interest
normalize counts in a combined_wig file (Pre-Processing->Normalize) - while most of the analysis methods automatically perform TTR normalization, and even the method for creating combined_wig files normalizes by default, one can choose to normalize a sample or whole dataset a different way (such as the Beta-Geometric Correction, BGC). But in most cases, users will not need to do an explicit normalization step.
make a scatter plot (Pre-Processing->Visuals->scatterplot) between 2 samples showing how correlated the mean insertion counts are at the gene level to check for consistency/reproducibility
make a correlation plot (Pre-Processing->Visuals->corrplot) among all samples to see which conditions appear more similar to each other, and to check that replicates are most highly correlated with each other (or check for outlier samples that do not correlate well with other replicates of the same condition, which might suggest they are bad or noisy)
Statistical Analysis Methods
The analysis methods available in Transit are divided into 3 categories:
Methods for analyzing single conditions or datasets and identifying essential genes
Gumbel method - looks for genes with larges ‘gaps’, or consecutive sequences of TA sites without insertions
Hidden Markov Model (HMM) - assigns genes to one of 4 states: ES (essential), GD (growth-defect), NE (non-essential), or GA (growth-advantaged), based on magnitudes of insertion counts (reflecting fitness effects of mutants)
Methods for pairwise comparisons of datasets (e.g. between a treatment and control condition) to identify conditionally essential genes
Resampling - a non-parametric test using permutations of insertion counts to simulate a null distribution of difference in mean counts for each gene
Mann-Whiney U-test - another non-parametric test based on comparison of rank-ordering of insertion counts in each gene
Methods for analyzing multiple conditions (>=2) to identify genes which exhibit significant variability of insertion counts across conditions
ZINB - similar to ANOVA, but uses the Zero-Inflated Negative Binomial disribution to model counts; this method has options for testing for interactions among covariates
Genetic Interaction analysis - this is a special case customized for testing interactions between 2 variables in a 2x2 experimental design (4 conditions)
Analyses for Tn5 Datasets
Although Transit was originally designed for analyzing Himar1 TnSeq datasets, many of the methods can be adapted for analyzing datasets that use other transposons like Tn5. The main difference is that the Himar1 transposon is restricted to insertions at TA dinucleotide sites, whereas Tn5 is capable of inserting more broadly at any coordinate in the genome.
1/3/2023: Previously (in Transit1 versions up through 3.2.7), we had a few statistical methods that were customized for analyzing Tn5 datasets. These have been temporarily disabled during the transition to Transit2. We will be adding back in the Tn5 analysis methods soon… (in a future release)
Results and Post-Processing
Most of the analysis methods in Transit generate output files that can be opened as spreadsheets in a program like Excel. The output files are generally tab-separated text files, with header lines demarcated by ‘#’ as prefixes.
For most of the analysis methods, the output file contains a row for each gene in the genome with information relevant to the statistical test, usually ending in columns labeled “P-value” and “Adj P-value” (Padj). The P-value is calculated based on the statistical test, and then all the P-values are adjusted by the Benjamini-Hochberg procedure to correct for multiple testing (aiming to limit the false discovery rate to FDR<5%).
In Transit, hits (or significant genes in the test) are generally defined as those with Padj<0.05, which can be identified in the output files by sorting on the “Adj P-value” column.
After the user runs a TRANSIT Analysis Method, various functions can be performed on the output file to better understand the results of the analyis performed. If using the GUI, the output file is visible in the Results Panel, along with a summary (params and outcomes). Click on the file and select one of the following (availablity depends on analysis that was run) from the action drop-down:
Display Table - an external window will appear in an spreadsheet-like format for you to view the file
Volcano Plot - an external window will appear that displays a plot of the LFCs vs. log10(pvalue) with a horizontal line indicating the thresold of significance
Display Heatmap - an external window will appear of clustered conditions and significant hits resulting from the analysis. This file will be also saved to your folder of choice and placed in the results pane, which then can be viewed by selection of the “View” option in the action dropdown.
Pathway Enrichment Analysis - this will search for significantly enriched pathways among the hits (Padj<0.05) in the selected file in the Results Panel (e.g. an output file from an analysis like resampling, ANOVA, etc)
Command Line
The analysis methods in Transit are also described in this PDF manual , focusing on command-line operations.
Most of the methods in Transit can be run from the command line. Typically, you run this as follows:
> python TRANSIT_PATH/src/transit.py <command> args...
Remember to use python3.
Commands include: gumbel, resampling, hmm, GI, tnseq_stats, anova… If you run ‘python TRANSIT_PATH/src/transit.py –help’, it will print out the full list of available commands.
> python TRANSIT_PATH/src/transit.py --help
The available subcommands are:
anova
gi
gumbel
hmm
...
The arguments would be whatever input files, options, and flags are appropriate for a given command. Generally, flags with “–” require an argument, whereas flags with “-” do not.
If you want a reminder of the usage for given command, use run that command without any arguments. For example:
> python3 TRANSIT_PATH/src/transit.py anova
Usage: python3 src/transit.py anova <combined_wig_file> <metadata_file> <annotation_file> <output_file> [Optional Arguments]
Optional Arguments:
--include-conditions <cond1,...> := Comma-separated list of conditions to use for analysis (Default: all)
--exclude-conditions <cond1,...> := Comma-separated list of conditions to exclude (Default: none)
--n <string> := Normalization method. Default: --n TTR
--ref <cond> := which condition(s) to use as a reference for calculating LFCs (comma-separated if multiple conditions)
--iN <N> := Ignore TAs within given percentage (e.g. 5) of N terminus. Default: --iN 0
--iC <N> := Ignore TAs within given percentage (e.g. 5) of C terminus. Default: --iC 0
--PC <N> := pseudocounts to use for calculating LFCs. Default: --PC 5
--alpha <N> := value added to mse in F-test for moderated anova (makes genes with low counts less significant). Default: --alpha 1000
-winz := winsorize insertion counts for each gene in each condition (replace max cnt with 2nd highest; helps mitigate effect of outliers)
Developers
Name |
Time Active |
Contact Information |
|---|---|---|
Thomas R. Ioerger |
2015-Present |
|
Michael A. DeJesus |
2015-2018 |
Rockefeller Univeristy (Bioinformatics Core) |
Chaitra Ambadipudi |
2015 |
|
Eric Nelson |
2016 |
|
Siddharth Subramaniyam |
2018 |
|
Jeff Hykin |
2022-2023 |
|
Sanjeevani Choudhery |
2021-2025 |
References
If you use TRANSIT, please cite the following reference:
Development of TRANSIT is funded by the National Institutes of Health (www.nih.gov/) grant U19 AI107774.
Other references for methods utilized by TRANSIT: