Pathway Enrichment Analysis
Pathway Enrichment Analysis provides a method to identify enrichment of functionally-related genes among those that are conditionally essential (i.e. significantly more or less essential between two conditions). The analysis is typically applied as post-processing step to the hits identified by a comparative analysis, such as resampling. Several analytical method are provided: Fisher’s exact test (FET, hypergeometric distribution), GSEA (Gene Set Enrichment Analysis) by Subramanian et al (2005), and Ontologizer. For Fisher’s exact test, genes in the resampling output file with adjusted p-value < 0.05 are taken as hits, and evaluated for overlap with functional categories of genes. The GSEA methods use the whole list of genes, ranked in order of statistical significance (without requiring a cutoff), to calculate enrichment.
Four systems of categories are provided for (but you can add your own): the Sanger functional categories of genes determined in the original annotation of the H37Rv genome (Cole et al, 1998, with subsequent updates), COG categories (Clusters of Orthologous Genes), also GO terms (Gene Ontology), and KEGG. Pre-formatted annotation files for M. tuberculosis H37Rv and several other mycobacteria can be found in pathways.html.
For other organisms, it might be possible to download COG categories from http://www.ncbi.nlm.nih.gov/COG/ and GO terms from http://www.geneontology.org or http://patricbrc.org. If these files can be obtained for your organism, they will have to be converted into the associations file format described below. (The pathways files for COG categories and GO terms in the Transit data directory should still work, because they just encode pathways names for all terms/ids.)
Pathway enrichment analysis is also available in the Transit GUI! (in Transit2) (see GUI Mode below)
Command Line Usage
Usage 1: # --M FET for Fisher's Exact Test (default)
> python3 transit.py pathway_enrichment <input_file> <associations> <pathways> <output_file> --M FET [Optional Arguments]
Optional Arguments:
--PC <int> := pseudo-counts to use in calculating p-value based on hypergeometric distribution. Default: --PC 2
--LFC-col <int> := column index (starting at 0) for LFC's
--focusLFC pos|neg := filter the output to focus on results with positive (pos) or negative (neg) LFCs (default: "all", no filtering)
--minLFC <float> := filter the output to include only genes that have a magnitude of LFC greater than the specified value (default: 0) (e.g. '--minLFC 1' means analyze only genes with 2-fold change or greater)
--qval <float> := filter the output to include only genes that have Qval less than to the value specified (default: 0.05)
--topk <int> := calculate enrichment among top k genes ranked by significance (Qval) regardless of cutoff (can combine with --focusLFC)
Usage 2: # GSEA for Gene Set Enrichment Method (Subramaniam et al, 2005)
> python3 transit.py pathway_enrichment <input_file> <associations> <pathways> <output_file> --M GSEA [Optional Arguments]
Optional Arguments:
--ranking <SLPV or LFC> := SLPV is signed-log-p-value, LFC is log2-fold-change from input. Default --ranking SLPV
--p <float> := exponent to use in calculating enrichment score; recommend trying 0 or 1 (as in Subramaniam et al, 2005)
--n-perm <int> := number of permutations to simulate for null distribution to determine p-value. Default --n-perm 10000
--LFC-col<int> := column index (starting at 0) for LFC's
Usage 3: # ONT for Ontologizer (Grossman et al, 2007)
> python3 transit.py pathway_enrichment <input_file> <associations> <pathways> <output_file> --M ONT [Optional Arguments]
Optional Arguments:
--LFC-col <int> := column index (starting at 0) for LFC's
Parameters
- Input File
The input file is the one obtained after using a comparitive analysis method in Transit (ANOVA, Resampling, GI, ZINB, Utest). GSEA method makes usage of the Adj P-value and Log 2 FC columns.
- Associations File
This is a tab-separated text file with 2 columns: pathway id, and pathway name. If a gene is in multiple pathways, the associated ids should be listed on separate lines. It is OK if there are no associations listed for some genes. Important: if pathways are hierarchical, you should expand this file to explicitly include associations of each gene with all parent nodes. Files with GO term associations will have to be pre-processed this way too.
Example: H37Rv_sanger_roles.dat
Rv3823c II.C.4
Rv3823c II.C
Rv3823c II
Rv0337c I.D.2
Rv0337c I.D
Rv0337c I
...
- Pathways File
This is a tab-separated text file with 2 columns: pathway id, and pathway name.
Example: sanger_roles.dat
I Small-molecule metabolism
I.A Degradation
I.A.1 Carbon compounds
I.A.2 Amino acids and amines
I.A.3 Fatty acids
I.A.4 Phosphorous compounds
...
- --M [FET|GSEA|ONT]
Methodology to be used. FET is used by default (even without specifying -M).
- --M FET
This implements Fisher’s Exact Test (hypergeometric distribution) to determine a p-value for each pathway, based on the proportion of pathway member observed in list of hits (conditionally essential gene by resampling, padj<0.05) compared to the background proportion in the overall genome, and p-values are adjusted post-hoc by the Benjamini-Hochberg procedure to limit the FDR to 5%.
In the output file, an “enrichment score” is reported, which is the ratio of the observed number of pathway members among the hits to the expected number. Pseudocounts of 2 are included in the calculation to reduce the bias toward small pathways with only a few genes; this can be adjusted with the --PC flag (below).
FET can be used with GO terms.
Additional flags for FET:
-focusLFC pos|neg : filter the output to focus on genes with positive (pos) or negative (neg) LFCs (default: “all”, no filtering)
-minLFC <float> : filter the output to include only genes that have |LFC| (magnitude of log2-fold change) >= the specified value (default: 0; e.g. ‘-minLFC 1’ means restriction to genes with 2-fold change or greater)
-qval <float> : set Q-value cutoff (analyze genes with Qval<cutoff) (default: 0.05)
-topk <int> : analyze enrichment in top K genes sorted by significance (Qval), regardless of Qval cutoff (can combine with -focusLFC)
-PC <int> : Pseudocounts used in calculating the enrichment score and p-value by hypergeometric distribution. Default: PC=2.
- --M GSEA
Gene Set Enrichment Analysis. GSEA assess the significance of a pathway by looking at how the members fall in the ranking of all genes. The genes are first ranked by significance from resampling. Specifically, they are sorted by signed-log-p-value, SLPV=sign(LFC)*(log(pval)), which puts them in order so that the most significant genes with negative LFC are at the top, the most significant with positive LFC are at the bottom, and insignificant genes fall in the middle. Roughly, GSEA computes the mean rank of pathway members, and evaluates significance based on a simulated a null distribution. p-values are again adjusted at the end by BH.
GSEA can be used with GO terms.
Additional flags for GSEA:
--ranking SLPV|LFC: method used to rank all genes; SLPV is signed-log-p-value (default); LFC is log2-fold-change from resampling
--p <float>: exponent to use in calculating enrichment score; recommend trying ‘--p 0’ (default) or ‘--p 1’ (as used in Subramaniam et al, 2005)
--Nperm <int>: number of permutations to simulate for null distribution to determine p-value (default=10000)
--LFC_col <int>: indicate column with log2FC (starting with 0; can also be negative, i.e. -1 means last col) (used for ranking genes by SLPV or LFC) (default: 6)
- --M ONT
Ontologizer is a specialized method for GO terms that takes parent-child relationships into account among nodes in the GO hierarchy. This can enhance the specificity of pathways detected as significant. (The problem is that there are many GO terms in the hierarchy covering similar or identical sets of genes, and often, if one node is significantly enriched, then several of its ancestors will be too, which obscures the results with redundant hits; Ontologizer reduces the significance of nodes if their probability distribution among hits can be explained by their parents.) Hierarhical relationships among GO terms are encoded in an OBO file, which is included in the src/pytransit/data/ directory.
For the ONT method in pathway_enrichment, the enrichment for a given GO term can be expressed (in a simplified way, leaving out the pseudocounts) as:
enrichment = log ( (b/q) / (m/p) )
where:
b is the number of genes with this GO term in the subset of hits (e.g. conditional essentials from resampling, with qval<0.05)
q is the number of genes in the subset of hits with a parent of this GO term
m is the total number of genes with this GO term in the genome
p is the number of genes in the genome with a parent of this GO term
So enrichment is the log of the ratio of 2 ratios:
the relative abundance of genes with this GO term compared to those with a parent GO term among the hits
the relative abundance of genes with this GO term compared to those with a parent GO term in the whole genome
Pathway Association Files
Pathway association files for several mycobacterial species (M. tuberculosis, M. smegmatis, M. abscessus, etc.) can be downloaded from our pathways.html web page. The pathway annotations include COG, KEGG, Sanger (Mtb only), and GO terms.
Note: The “Sanger” roles are custom pathway associations for M. tuberculosis defined in the original Nature paper on the H37Rv genome sequence (Cole et al., 1998) (Table 1). They are more specific that COG categories, but less specific than GO terms. For other organisms, one should be able to find GO terms (e.g. on PATRIC, Uniprot, or geneontology.org) and COG roles (from https://ftp.ncbi.nih.gov/pub/COG/COG2020/data/, (Galerpin et al, 2021) ).
Here are the recommended combinations of pathway methods to use for different systems of functional categories:
For COG, use ‘-M FET’
For KEGG and Sanger pathways, try both FET and GSEA
For GO terms, use ‘M -ONT’
Examples
# uses Fisher's exact test by default (with PC=2 as pseudocounts)
> python3 transit.py pathway_enrichment resampling_glyc_chol.txt $DATA/H37Rv_sanger_roles.dat $DATA/sanger_roles.dat pathways_glyc_chol_Sanger.txt
# can do this with GO terms too
> python3 transit.py pathway_enrichment resampling_glyc_chol.txt $DATA/H37Rv_GO_terms.txt $DATA/GO_term_names.dat pathways_glyc_chol_GO.txt
# with COG_20 categories
> python3 transit.py pathway_enrichment resampling_glyc_chol.txt $DATA/Mycobacterium_tuberculosis_H37Rv_COG_20_roles.associations.txt $DATA/COG_20_roles.txt pathways_glyc_chol_COG.txt
# can also do GSEA method (on any system of functional categories)
> python3 transit.py pathway_enrichment resampling_glyc_chol.txt $DATA/H37Rv_sanger_roles.dat $DATA/sanger_roles.dat pathways_Sanger_GSEA.txt --M GSEA
# Ontologizer is a specialized method for GO terms
> python3 transit.py pathway_enrichment resampling_glyc_chol.txt $DATA/H37Rv_GO_terms.txt $DATA/GO_term_names.dat pathways_Ontologizer.txt --M ONT
The $DATA environment variable in these examples refers to the Transit data directory, e.g. src/pytransit/data/.
GUI Mode
Pathway Enrichment can be accessed from the “Post-Processing” tab in the Menu Bar (“1.” in figure below) of through the actions dropdown of a valid results file in the results panel (“2.” in figure below).

The parameters to input through the parameter panel for the method is equivalent to the command line usage, except in the GUI format we have pre-set some of the common Pathway Systems for ease of the user.

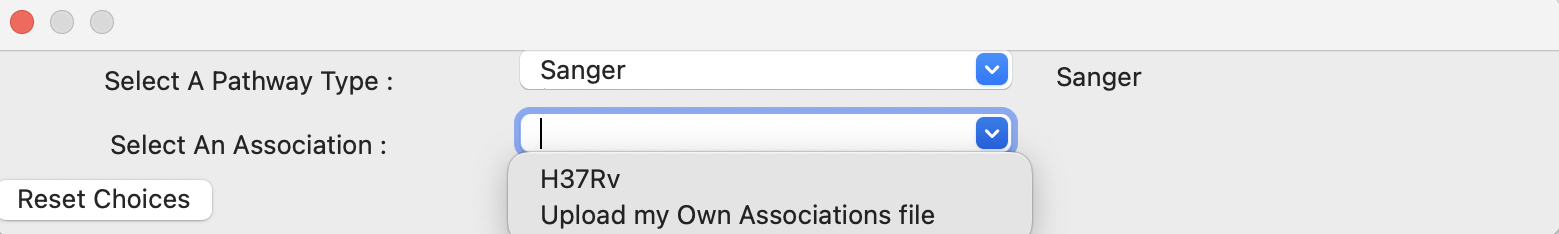
- Select Pathway System Button
This button allows you to select from a set of pre-loaded pathway systems or upload your own. Each of the dropdowns populates based on the selection of the other. For example, if M.Smegmatis is selected as the organism of interest (Association), the pathways to select from will be COG_20 and GO along with an option for the user to upload their own.
Output and Diagnostics
All output files contain the following columns:
Column Name |
Column Description |
|---|---|
Pathway |
The pathways of interest using the pathway file selected |
Pathway Description |
Description of the Pathway of interest |
Number of Genes in Path |
Number of Total Genes in the Pathway, using the associations file selected |
Enrichment Score |
Enrichment Score of the Pathway |
P Value |
P Value Determined by the Pathway Enrichment Analysis Method slected |
Adj P Value |
FDR-corrected P Value |
Relevant Genes |
In the output files from FET and ONT, these genes are those in the siginificant
genes in path column, the overlap of the pathway and the significant genes from
the input file (Adj P Value < 0.05). Since GSEA looks at the ranking of genes in
a pathway using the entire genome, the genes in this column are GSEA calculated
hits in the pathway.
|
The files are typically sorted by significance (Adj P Value) or Enrichment Score. There are additional columns in the output files relating to the method used to conduct pathway enrichment. For example, GSEA also contains a mean rank column which could be useful to sort by.
Run-time
A typical run of the pathway enrichment method takes less than 1 minute.